Мутации де ново. Детекция мутации de novo в гене дистрофина и её значение для медико-генетического консультирования при мышечной дистрофии Дюшенна (клиническое наблюдение)
Медицинская цитогенетика - изучение кариотипа человека в норме и при патологии. Это направление возникло в 1956 г., когда Тио и Леван усовершенствовали метод приготовления препаратов метафазных хромосом и впервые установили модальное число хромосом (2n=46) в диплоидном наборе. В 1959 г. была расшифрована хромосомная этиология ряда заболеваний - синдромов Дауна, Клайнфельтера, Шерешевского-Тернера и некоторых других синдромов аутосомных трисомий. Дальнейшее развитие медицинской цитогенетики в конце 1960-х годов было обусловлено появлением методов дифференциального окрашивания метафазных хромосом, дающих возможность идентификации хромосом и их отдельных районов. Методы дифференциального окрашивания не всегда обеспечивали правильность установления точек разрывов в результате структурных перестроек хромосом. В 1976 г. Юнис разработал новые методы их изучения на стадии прометафазы, которые получили название «высокоразрешающие методы».
Использование таких методов позволило получить хромосомы с разным количеством сегментов (от 550 до 850) и дало возможность проведения идентификации нарушений с вовлечением небольших их участков (микроперестроек). С начала 1980-х гг. цитогенетика человека вступила в новый этап развития: в практику был внедрен хромосомный анализ молекулярно-цитогенетических методов, флюоресцентной гибридизации in situ (FISH - Fluorescence In Situ Hybridization). Этот метод широко используют для выявления более тонких структурных аномалий хромосом, которые неразличимы при дифференциальном окрашивании. В настоящее время применение различных методов хромосомного анализа позволяет успешно проводить пре- и постнатальную диагностику хромосомных болезней.
Хромосомные болезни - большая группа клинически многообразных состояний, характеризуемых множественными врожденными пороками развития, этиология которых связана с количественными или структурными изменениями кариотипа.
В настоящее время различают почти 1000 хромосомных аномалий, из них более 100 форм имеют клинически очерченную картину и называются синдромами; их вклад в спонтанные аборты, неонатальную смертность и заболеваемость весьма значителен. Распространенность хромосомных аномалий среди спонтанных абортов составляет в среднем 50%, среди новорожденных с грубыми множественными врожденными пороками развития - 33%, мертворожденных и перинатально умерших с врожденными пороками развития - 29%, недоношенных с врожденными пороками развития - 17%, новорожденных с врожденными пороками развития - 10%, мертворожденных и перинатально умерших - 7%, недоношенных - 2,5%, всех новорожденных - 0,7%.
Большинство хромосомных болезней являются спорадическими, возникающими заново вследствие геномной (хромосомной) мутации в гамете здорового родителя или в первых делениях зиготы, а не наследуемыми в поколениях, что связано с высокой смертностью больных в дорепродуктивном периоде. Фенотипическую основу хромосомных болезней составляют нарушения раннего эмбрионального развития. Именно поэтому патологические изменения складываются еще в пренатальном периоде развития организма и либо обусловливают гибель эмбриона или плода, либо создают основную клиническую картину заболевания уже у новорожденного (исключение составляют аномалии полового развития, формирующиеся в основном в период полового созревания). Раннее и множественное поражение систем организма характерно для всех форм хромосомных болезней. Это черепно-лицевые дизморфии, врожденные пороки развития внутренних органов и частей тела, замедленные внутриутробный и постнатальный рост и развитие, отставание психического развития, пороки центральной нервной системы, сердечно-сосудистой, дыхательной, мочеполовой, пищеварительной и эндокринной систем, а также отклонения в гормональном, биохимическом и иммунологическом статусе. Для каждого хромосомного синдрома характерен комплекс врожденных пороков развития и аномалий развития, присущий в какой-то мере только данному типу хромосомных патологий. Клинический полиморфизм каждой хромосомной болезни в общей форме обусловлен генотипом организма и условиями среды. Вариации в проявлениях патологии могут быть очень широкими - от летального эффекта до незначительных отклонений в развитии. Несмотря на хорошую изученность клинических проявлений и цитогенетики хромосомных болезней, их патогенез даже в общих чертах еще не ясен. Не разработана общая схема развития сложных патологических процессов, обусловленных хромосомными аномалиями и приводящих к появлению сложнейших фенотипов хромосомных болезней.
Основные типы хромосомных аномалий
Все хромосомные болезни по типу мутаций можно разделить на две большие группы: вызванные изменением числа хромосом при сохранении структуры последних (геномные мутации) и обусловленные изменением структуры хромосомы (хромосомные мутации). Геномные мутации возникают вследствие нерасхождения или утраты хромо-сом в гаметогенезе или на ранних стадиях эмбриогенеза. У человека обнаружено только три типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия. Частота возникновения триплоидных (Зn=69) и тетраплоидных (4n=92) мутаций очень низка, в основном их обнаруживают среди спонтанно абортированных эмбрионов или плодов и у мертворожденных. Продолжительность жизни новорожденных с такими нарушениями - несколько дней. Геномные мутации по отдельным хромосомам многочисленны, они составляют основную массу хромосомных болезней. При этом из всех вариантов анеуплоидий встречаются только трисомии по аутосомам, полисомии по половым хромосомам (три-, тетра- и пентасомии), а из моносомий встречается только моносомия X.
Полные трисомии или моносомии переносятся организмом тяжелее, чем частичные, дисбаланс по крупным хромосомам встречается у живорожденных значительно реже, чем по мелким. Полные формы хромосомных аномалий вызывают значительно более серьезные отклонения, чем мозаичные. Аутосомные моносомии среди живорожденных очень редки, это мозаичные формы с большой долей нормальных клеток. Доказан факт относительно малой генетической ценности гетерохроматиновых районов хромосом. Именно поэтому полные трисомии у живорожденных наблюдают по тем аутосомам, которые богаты гетерохроматином, - 8, 9, 13, 14, 18, 21, 22 и X. Этим объясняется хорошая переносимость пациентами даже тройной дозы материала Y-хромосомы и почти полная утрата длинного ее плеча. Совместимую с постнатальной жизнью полную моносомию по Х-хромосоме, приводящую к развитию синдрома Шерешевского-Тернера, а также тетра- и пентасомии наблюдают только по Х-хромосоме, которая гетерохроматизирована.
Хромосомные мутации, или структурные хромосомные перестройки, - нарушения кариотипа, сопровождаемые или не сопровождаемые дисбалансом генетического материала в пределах одной или нескольких хромосом (внутри- и межхромосомные перестройки).
В подавляющем большинстве случаев структурные хромосомные мутации пере-дает потомству один из родителей, в кариотипе которого присутствует сбалансированная хромосомная перестройка. К ним относят реципрокную (взаимную) сбалансированную транслокацию без потери участков вовлеченных в нее хромосом. Она, как и инверсия, не вызывает патологических явлений у носителя. Однако при образовании гамету носителей сбалансированных транслокаций и инверсий могут образовываться несбалансированные гаметы. Робертсоновская транслокация - транслокация между двумя акроцентрическими хромосомами с потерей их коротких плеч - приводит к образованию одной метацентрической хромосомы вместо двух акроцентрических. Носители такой транслокации здоровы, потому что потеря коротких плеч двух акроцентрических хромосом компенсируется работой таких же генов в остальных 8 акроцентрических хромосомах. При созревании половых клеток случайное распределение (при клеточных делениях) двух перестроенных хромосом и их гомологов приводит к появлению нескольких типов гамет, одни из которых нормальны, другие содержат такую комбинацию хромосом, которая при оплодотворении дает зиготу со сбалансированным перестроенным кариотипом, третьи дают при оплодотворении хромосомно несбалансированные зиготы.
При несбалансированном хромосомном наборе (делеции, дупликации, инсерции) у плода развиваются тяжелые клинические патологии, как правило, в виде комплекса врожденных пороков развития. Недостаток генетического материала вызывает более серьезные пороки развития, чем его избыток.
Значительно реже структурные аберрации возникают de novo. Родители пациента с хромосомной болезнью обычно кариотипически нормальны. Хромосомная болезнь в этих случаях возникает de novo в результате передачи от одного из родителей геномной или хромосомной мутации, возникшей однократно в одной из гамет, или такая мутация возникает уже в зиготе. Это не исключает повторного возникновения хромосомного нарушения у детей в данной семье. Есть семьи, предрасположенные к повторным случаям нерасхождения хромосом. Мутациями, возникшими de novo, являются почти все случаи известных полных трисомий и моносомий. Основной механизм возникновения структурной перестройки любого типа - разрыв в одной или нескольких хромосомах с последующим воссоединением образовавшихся фрагментов.
Клинические показания к цитогенетической диагностике
Цитогенетический метод исследования занимает ведущее место среди методов лабораторной диагностики при медико-генетическом консультировании и в пре-натальной диагностике. Однако следует строго придерживаться объективных
показаний для направления пациентов на исследование кариотипа.
Основные показания к пренатальной диагностике:
хромосомная аномалия у предыдущего ребенка в семье;
мертворожденный ребенок с хромосомной аномалией;
хромосомные перестройки, хромосомный мозаицизм или анеуплоидия по половым хромосомам у родителей;
результаты исследования сыворотки крови у матери, указывающие на повы-шенный риск хромосомной аномалии у плода (группа риска);
возраст матери;
выявленные при ультразвуковом исследовании аномалии плода;
подозрение на мозаицизм у плода при предыдущем цитогенетическом исследовании;
подозрение на синдром с хромосомной нестабильностью.
Исследование кариотипа при постнатальной диагностике рекомендуют проводить при наличии у пациента:
первичной или вторичной аменореи или ранней менопаузы;
аномальной спермограммы - азооспермии или выраженной олигоспермии;
клинически выраженных отклонений в росте (низкий, высокий рост) и размерах головы (микро-, макроцефалия);
аномальных гениталий;
аномального фенотипа или дизморфий;
врожденных пороков развития;
умственной отсталости или нарушений развития;
проявлений делеционного/микроделеционного/дупликационного синдрома;
Х-сцепленного рецессивного заболевания у женщин;
клинических проявлений синдромов хромосомной нестабильности;
при мониторинге после трансплантации костного мозга.
Цитогенетические исследования следует провести у супружеской пары:
при хромосомных аномалиях или необычных вариантах хромосом у плода, обнаруженных при пренатальной диагностике;
повторных выкидышах (3 и более); мертворождении, неонатальной смерти плода, невозможности обследования пораженного плода;
наличии у ребенка хромосомной аномалии или необычного хромосомного варианта;
бесплодии неизвестной этиологии.
Показанием к цитогенетическому исследованию является наличие у родственников пациента:
хромосомных перестроек;
умственной отсталости предположительно хромосомного происхождения;
репродуктивных потерь, врожденных пороков развития плода или мертворождения неясного происхождения.
Показания к исследованию FISH-методом:
подозрение на микроделеционный синдром, для которого доступна молекулярно-цитогенетическая диагностика (наличие соответствующих ДНК-зондов);
повышенный риск микроделеционного синдрома по анамнестическим данным;
клинические признаки, позволяющие предположить мозаицизм по опреде-ленному хромосомному синдрому;
состояния после трансплантации костного мозга, когда донор и реципиент разного пола;
подозрение на хромосомную аномалию при стандартном цитогенетическом исследовании, когда FISH-метод может быть полезным для дальнейшего
уточнения характера аномалии, или в ситуациях, когда имеются характерные клинические проявления;
наличие сверхчисленной маркерной хромосомы;
подозрение на скрытую хромосомную перестройку.
FISH-метод при анализе метафаз показан:
при маркерных хромосомах;
дополнительном материале неизвестного происхождения на хромосоме;
хромосомных перестройках;
подозрении на потерю хромосомного сегмента;
мозаицизме.
FISH-метод при анализе интерфазных ядер показан:
при численных хромосомных аномалиях;
дупликациях;
делениях;
перестройках хромосом;
определении хромосомного пола;
амплификации генов.
Методы цитогенетического исследования
:
Исследование и описание характерных особенностей метафазных хромосом особенно важны для практической цитогенетики. Отдельные хромосомы в пределах группы распознают с помощью методов дифференциального окрашивания. Эти методы позволяют обнаруживать неоднородность структуры хромосомы по длине, определяемую особенностями комплекса основных молекулярных компонентов хромосом - ДНК и белков. Проблема распознавания индивидуальных хромосом в кариотипе важна для развития цитогенетической диагностики хромосомных болезней у человека.
Методы цитогенетического исследования делятся на прямые и непрямые. Прямые методы применяют в тех случаях, когда нужен быстрый результат и имеется возможность получить препараты хромосом клеток, делящихся в организме. Непрямые методы включают в качестве обязательного этапа более или менее длительное культивирование клеток в искусственных питательных средах. Промежуточное положение занимают методы, включающие кратковременное культивирование (от нескольких часов до 2-3 сут).
Основной объект цитогенетического исследования прямыми и непрямыми методами - стадия метафазы митоза и различные стадии мейоза. Метафаза мито¬за служит основным предметом цитогенетического исследования, так как именно на этой стадии возможны точная идентификация хромосом и выявление их ано¬малий. Хромосомы в мейозе исследуют для обнаружения некоторых типов пере¬строек, по природе своей не обнаруживаемых в метафазе митоза.
Биологический материал для цитогенетических исследований. Обработка клеточных культур. Приготовление хромосомных препаратов
В качестве материала для получения хромосом человека и их исследования могут быть использованы клетки любой ткани, доступной для биопсии. Чаще всего используют периферическую кровь, кожные фибробласты, костный мозг, клетки амниотической жидкости, ворсинчатого хориона. Наиболее доступны для исследования хромосом лимфоциты периферической крови человека.
В настоящее время практически во всех лабораториях мира для постановки культуры лимфоцитов применяют метод с использованием цельной периферической крови. Кровь в количестве 1-2 мл заранее берут из локтевой вены в стерильную пробирку или флакон с раствором гепарина. Во флаконе кровь можно хранить 24-48 ч в холодильнике при температуре 4-6 °С. Постановку культуры лимфоцитов осуществляют в специальном боксовом помещении или в рабочей комнате под ламинарным шкафом в стерильных условиях. Такие условия обязательны для предотвращения заноса в культуру крови патогенной флоры. Если есть подозрение на загрязненность крови или другого материала, необходимо в культуральную смесь добавить антибиотики. Флаконы с культуральной смесью инкубируют в термостате при температуре +37 °С в течение 72 ч (идет активный рост и деление клеток). Основное назначение методических приемов при обработке клеточных культур и приготовлении из них хромосомных препаратов - получить на препарате достаточное количество метафазных пластинок с таким разбросом хромосом, при котором можно оценить длину, форму и другие морфологические признаки каждой хромосомы набора.
Накопление клеток в метафазе митоза и получение на препарате качественных пластинок происходит с помощью ряда последовательных процедур:
колхинизации - воздействия на клетки цитостатиками колхицином или колцемидом, блокирующими митоз в стадии метафазы;
гипотонизации культур;
фиксации клеток смесью метилового спирта с уксусной кислотой;
нанесения клеточной взвеси на предметное стекло.
Колхинизацию культур клеток осуществляют за 1,5-2 ч до начала фиксации. После введения колхицина флаконы с клеточными культурами продолжают инкубировать в термостате. По окончании инкубации культуральную смесь из каждого флакона сливают в чистые центрифужные пробирки и подвергают центрифугиро-ванию. Затем к осадку клеток добавляют гипотонический раствор калия хлорида, предварительно нагретый до температуры +37 °С.
Гипотонизацию проводят в термостате при температуре +37 °С в течение 15 мин. Гипотонический раствор KCI способствует лучшему разбросу хромосом на предметном стекле. После гипотонизации клетки переводят в осадок центрифуги-рованием и подвергают фиксации. Фиксацию проводят смесью метилового (или этилового) спирта с уксусной кислотой.
Завершающий этап - приготовление хромосомных препаратов для получения хорошо «распластанных» метафазных пластинок с сохранением целостности, полноты хромосомного набора в каждой из них. На мокрые, охлажденные пред-метные стекла наносят клеточную взвесь, после чего стекла высушивают при ком-натной температуре и маркируют.
Методы дифференциального окрашивания хромосом
С 1971 г. в цитогенетике нашли широкое распространение методы, позволяющие дифференциально окрашивать каждую хромосому набора по ее длине. Практическое значение этих методов состоит в том, что дифференциальная окраска позволяет идентифицировать все хромосомы человека благодаря специфическому рисунку продольной окрашиваемости для каждой хромосомы. Для окраски может быть пригодна любая краска, состоящая из основного красителя, поскольку главным красящим субстратом хромосом является комплекс ДНК с белками. В практике цитогенетических исследований наибольшее применение получили следующие методы.
G-метод окраски - самый распространенный метод из-за простоты, надежности и доступности необходимых реактивов. После окраски каждая пара хромосом приобретает исчерченность по длине благодаря чередованию по-разному окрашенных гетерохроматиновых (темных) и эухроматиновых (светлых) сегментов, которые принято обозначать как G-сегменты. С-метод окраски обеспечивает выявление лишь некоторых районов хромосом. Это районы гетерохроматина, локализованного в околоцентромерных участках длинных плеч хромосом 1, 9 и 16 и в длинном плече Y-хромосомы, а также в коротких плечах акроцентрических хромосом. R-метод окраски препаратов хромосом показывает картину дифференциальной сегментации, обратной G-методу. Этим методом хорошо прокрашиваются дистальные сегменты хромосом, что очень важно при идентификации мелких пере-строек с вовлечением концевых участков. Q-метод окраски обеспечивает дифференциальную флюоресцентную окраску индивидуальных хромосом набора, позволяет идентифицировать каждую пару гомологов, а также определить наличие Y-хромосомы в интерфазных ядрах по свечению тельца Y-хроматина.
Принципы хромосомного анализа
Обязательным этапом исследования является визуальный анализ хромосом под микроскопом с использованием тысячекратного увеличения (х1000) при окулярах х10 и иммерсионном объективе х100. Оценку качества и пригодности хромосомных препаратов для исследования, а также отбор метафазных пластинок для анализа проводят при малом увеличении (х100). Для исследования выбирают хорошо окрашенные, полные метафазные пластинки с хорошим разбросом хромосом. Исследователь подсчитывает общее количество хромосом и проводит оценку структуры каждой хромосомы путем сопоставления исчерченности гомологов, а также сопоставления наблюдаемой картины с цитогенетическими картами (схемами) хромосом.
Использование компьютерных систем анализа изображений существенно облегчает задачу цитогенетика, повышает качество его работы и предоставляет возможность быстрого и простого документирования результатов исследования. Для обеспечения высокого качества работы рекомендуют участие двух специалистов в проведении цитогенетического исследования каждого образца. Документом, подтверждающим исследование, служит протокол, в котором указывают координаты просмотренных клеток, количество хромосом в каждой из них, обнаруженные перестройки, формулу кариотипа и заключение, а также фамилию пациента, дату и номер исследования, фамилию и подпись врача (врачей), проводившего исследование. Следует сохранять препараты и изображения хромосом для последующего просмотра.
ОСНОВНЫЕ ПРАВИЛА ОПИСАНИЯ ХРОМОСОМНЫХ АНОМАЛИЙ СОГЛАСНО МЕЖДУНАРОДНОЙ СИСТЕМЕ ЦИТОГЕНЕТИЧЕСКОЙ НОМЕНКЛАТУРЫ
Запись формулы кариотипа необходимо проводить в соответствии с действую-щей версией Международной системы цитогенетической номенклатуры человека - International System for human Cytogenetic Nomenclature. Ниже рассмотрены аспекты применения номенклатуры, которые наиболее часто встречаются в клинической цитогенетической практике.
Количество и морфология хромосом:
В кариотипе хромосомы разделяют на семь легкоразличимых групп (А-G) в соответствии с их размером и положением центромеры. Аутосомы - это хромосо¬мы с 1-й по 22-ю, половые хромосомы - X и Y.
Группа А (1-3) - большие метацентрические хромосомы, которые можно отличить друг от друга по размеру и положению центромеры.
Группа В (4-5) - большие субметацентрические хромосомы.
Группа С (6-12, X) - метацентрические и субметацентрические хромосомы среднего размера. Х-хромосому относят к самым крупным хромосомам в этой группе.
Группа D (13-15) - акроцентрические хромосомы среднего размера со спутниками.
Группа Е (16-18) - относительно небольшие метацентрические и субметацентрические хромосомы.
Группа F (19-20) - маленькие метацентрические хромосомы.
Группа G (21-22, Y) - маленькие акроцентрические хромосомы со спутниками. Y-хромосома не имеет спутников.
Каждая хромосома состоит из непрерывного ряда полос, которые размещаются по длине плеч хромосом в строго ограниченных районах (участках). Хромосомные районы специфичны для каждой хромосомы и имеют существенное значение для их идентификации. Полосы и районы нумеруют в направлении от центромеры к теломере по длине каждого плеча. Районы - участки хромосомы, расположенные между двумя соседними полосами. Для обозначения коротких и длинных плеч хромосом используют следующие символы: р - короткое плечо и q - длинное плечо. Центромера (сеп) обозначена символом 10, часть центромеры, прилежащая к короткому плечу, - р10, к длинному плечу - q10. Район, ближайший к центромере, обозначают цифрой 1, следующий район - цифрой 2 и т. д.
Для обозначения хромосом используют четырехзначную символику:
1-й символ - номер хромосомы;
2-й символ (р или q) - плечо хромосомы;
3-й символ - номер района (участка);
4-й символ - номер полосы в пределах этого района.
Например, запись 1р31 указывает на хромосому 1, ее короткое плечо, район 3, полосу 1. Если полоса подразделяется на субполосы, после обозначения полосы ставят точку, затем пишут номер каждой субполосы. Субполосы, так же как и полосы, нумеруют в направлении от центромеры к теломере. Например, в полосе 1р31 выделяют три субполосы: 1р31.1, 1р31.2 и 1р31.3, из которых субполоса 1р31.1 проксимальна по отношению к центромере, а субполоса 1р31.3 - дистальна. Если субполосы подразделяют дополнительно на части, их нумеруют цифрами без пунктуации. Например, субполоса 1р31.1 делится на 1р31.11,1р31.12 и т. д.
ОБЩИЕ ПРИНЦИПЫ ОПИСАНИЯ НОРМАЛЬНОГО И АНОМАЛЬНОГО КАРИОТИПА
В описании кариотипа первым пунктом указывают общее количество хромосом, включая половые хромосомы. Первое число отделяют от остальной части записи запятой, затем записывают половые хромосомы. Аутосомы обозначают только в случае аномалий.
Нормальный человеческий кариотип выглядит следующим образом:
46,XX - нормальный кариотип женщины;
46,XY - нормальный кариотип мужчины.
При хромосомных аномалиях первыми записывают аномалии половых хромо-сом, затем аномалии аутосом в порядке возрастания номеров и независимо от типа аномалии. Каждую аномалию отделяют запятой. Для описания структурно перестроенных хромосом используют буквенные обозначения. Хромосому, вовлеченную в перестройку, записывают в круглых скобках после символа, обозначающего тип перестройки, например: inv(2), del(4), r(18). Если в перестройке участвуют две или более хромосом, между обозначениями номера каждой из них ставят точку с запятой (;).
Знаки (+) или (-) ставят перед хромосомой для обозначения аномалии с указанием дополнительной или отсутствующей хромосомы (нормальной или аномальной), например: +21,-7,+der(2). Их также используют для обозначения уменьшения или увеличения длины плеча хромосомы после символа (р или q); с этой целью указанные выше знаки можно применять только в тексте, но не в описании кариотипа, например: 4р+, 5q-. При описании размеров гетерохроматиновых сегментов, спутников и спутнич- ных нитей знак (+) (увеличение) или (-) (уменьшение) ставят непосредственно за обозначением соответствующего символа, например: 16qh+, 21ps+, 22pstk+. Знак умножения (х) используют для описания множественных копий перестроенных хромосом, но его нельзя использовать для описания множественных копий нормальных хромосом, например: 46,XX,del(6)(q13q23)х2. Для указания альтернативных интерпретаций аномалий используют символ (оr), например: 46,XX,del(8)(q21.1) or i(8)(p10).
Кариотипы разных клонов разделяют косой чертой (/). Квадратные скобки ставят после описания кариотипа, для обозначения абсолютного количества клеток в данном клоне. Для того чтобы указать причину возникновения разных клонов, используют символы mos (мозаицизм - клеточные линии произошли из одной зиготы) и chi (химера - клеточные линии произошли из разных зигот), которые приводят перед описанием кариотипа. При перечислении кариотипов нормальный диплоидный клон всегда указывают последним, например: mos47,XY,+21/46,XY; mos47,XXY/46,XY.
Если есть несколько аномальных клонов, запись проводят в порядке увеличе¬ния их размера: первый - наиболее часто встречаемый, затем по нисходящей. Самым последним указывают нормальный клон, например: mos45,X/47,XXX /46,ХХ. Аналогичную запись используют и в кариотипе, имеющем два нормальных клона, например: chi46,XX/46,XY. Если в кариотипе присутствуют два аномальных клона, один из которых имеет числовую аномалию, а другой - структурную перестройку, то клон с числовой аномалией записывают первым. Например: 45,X/46,X,i(X)(q10).
Когда оба клона имеют числовые аномалии, сначала записывают клон, имеющий аутосому с меньшим порядковым номером, например: 47,ХХ,+8/47,ХХ,+21; клон с аномалиями половых хромосом всегда ставят первым, например: 47,ХХХ/47,ХХ,+21 .
Тот факт, что кариотип является гаплоидным или полиплоидным, будет очеви-ден из числа хромосом и дальнейших обозначений, например: 69,XXY. Все измененные хромосомы должны быть обозначены относительно соответствующего уровня плоидности, например: 70,XXY,+21.
Материнское или отцовское происхождение аномальной хромосомы обозначают символами mat и pat соответственно после описываемой аномалии, например: 46,XX,t(5;6)(q34;q23)mat,inv(14)(q12q31)pat; 46,XX,t(5;6)(q34;q23)mat,inv(14) (q12q31)mat. Если известно, что хромосомы родителей нормальны по сравнению с данной аномалией, ее рассматривают как вновь возникшую и обозначают символом denovo (dn), например: 46,XY,t(5;6)(q34;q23)mat,inv (14)(q12q31)dn.
Описание численных аномалий хромосом:
Знак (+) или (-) ставят для обозначения потери или приобретения дополни-тельной хромосомы при описании численных аномалий.
47,XX,+21 - кариотип с трисомией 21.
48,XX,+13,+21 - кариотип с трисомией 13 и трисомией 21.
45,XX,-22 - кариотип с моносомией 22.
46,XX,+8,-21 - кариотип с трисомией 8 и моносомией 21.
Исключением из этого правила являются конституциональные аномалии поло-вых хромосом, которые записывают без использования знаков (+) и (-).
45,X - кариотип с одной Х-хромосомой (синдром Шерешевского-Тернера).
47,XXY - кариотип с двумя Х-хромосомами и одной Y-хромосомой (синдром Клайнфельтера).
47,XXX - кариотип с тремя Х-хромосомами.
47,XYY - кариотип с одной Х-хромосомой и двумя Y-хромосомами.
48,XXXY - кариотип с тремя Х-хромосомами и одной Y-хромосомой.
Описание структурных аномалий хромосом
В описании структурных перестроек используют как краткую, так и детальную системы записи. При использовании краткой системы указывают только тип хромосомной перестройки и точки разрыва. Записывают тип хромосомной аномалии, хромосому, вовлеченную в данную аномалию, и в круглых скобках - точки разры-ва. Краткая система не дает возможности однозначного описания сложных хромосомных перестроек, которые иногда выявляют при анализе кариотипов опухолей.
Краткая система обозначения структурных перестроек
Если в перестройку, возникшую в результате двух разрывов, произошедших в одной хромосоме, вовлечены оба плеча, точку разрыва в коротком плече записыва¬ют перед точкой разрыва в длинном плече: 46,XX,inv(2)(p21q31). Когда две точки разрыва находятся в одном плече хромосомы, первой указывают проксимальную к центромере точку разрыва: 46,XX,inv(2)(p13p23). В случае, когда в перестройку вовлечены две хромосомы, первой указывают либо хромосому с меньшим поряд-ковым номером, либо половую хромосому: 46,XY,t(12;16)(q13;p11.1); 46,X,t(X;18) (p11.11;q11.11).
Исключением из правила являются перестройки с тремя точками разрыва, когда фрагмент одной хромосомы вставляется в район другой хромосомы. При этом хромосому-реципиента записывают первой, а хромосому-донора последней, даже если это половая хромосома или хромосома с меньшим порядковым номером: 46,X,ins(5;X)(p14;q21q25); 46,XY,ins(5;2)(p14;q22q32). Если перестройка затрагивает одну хромосому, первыми указывают точки разрыва в сегменте, где образовалась вставка. В случае прямой инсерции первой записывают проксимальную к центромере точку разрыва вставленного фрагмента, а затем - дистальную точку разрыва. При инвертированной вставке - наоборот.
Для обозначения транслокаций, в которые вовлечены три разные хромосомы, на первом месте указывают половую хромосому или хромосому с меньшим поряд¬ковым номером, затем хромосому, получившую фрагмент от первой хромосомы, и, наконец, хромосому, отдавшую фрагмент первой хромосоме. 46,XX,t(9;22;17) (q34;q11.2;q22) - фрагмент хромосомы 9, соответствующий дистальному району 9q34, перенесен на хромосому 22, в сегмент 22q11.2, фрагмент хромосомы 22, соответствующий дистальному району 22q11.2, перенесен на хромосому 17, в сегмент 17q22, а фрагмент хромосомы 17, соответствующий дистальному району 17q22, перенесен на хромосому 9, в сегмент 9q34.
Детальная система обозначения структурных перестроек. В соответствии с детальной системой обозначений структурные перестройки хромосом определяют по составу полос в них. Все обозначения, употребляемые в краткой системе, сохраняются и в детальной системе. Однако в детальной системе приводят подробное описание состава полос в перестроенных хромосомах с примене¬нием дополнительных символов. Двоеточие (:) обозначает точку разрыва, а двойное двоеточие (::) - разрыв с последующим воссоединением. Стрелкой (->) указывается направление переноса фрагментов хромосомы. Концы плеч хромосом обозначают символом ter (терминальный), pter или qter означают конец короткого или длинного плеча соответственно. Символ сеп используют для обозначения центромеры.
Типы хромосомных перестроек
Дополнительный материал неизвестного происхождения. Символ add (от лат. additio - прибавление) используют для указания на допол-нительный материал неизвестного происхождения, присоединившийся к хромосомному району или полосе. Дополнительный материал, присоединившийся к терминальному участку, будет вызывать увеличение длины плеча хромосомы. При описании хромосом с дополнительным материалом неизвестного проис-хождения в обоих плечах перед номером хромосомы ставят символ der. Если неизвестный дополнительный материал вставлен в плечо хромосомы, для описания используют символы ins и (?).
Делеции. Символ del используют для обозначения терминальных (концевых) и интерстициальных делеций:
46,XX,del(5)(q13)
46,XX,del (5) (pter->q13:)
Знак (:) означает, что разрыв произошел в полосе 5q13, в результате хромосома 5 состоит из короткого плеча и части длинного плеча, заключенной между центро-мерой и сегментом 5q13.
46,XX,del(5)(q13q33)
46,XX,del(5)(pter->q13::q33->qter)
Знак (::) означает разрыв и воссоединение полос 5ql3 и 5q33 длинного плеча хромосомы 5. Сегмент хромосомы между этими полосами делетирован.
Производные, или дериватные, хромосомы (der) - это хромосомы, возникшие в результате перестроек, затрагивающих две и более хромосом, а также в результате множественных перестроек внутри одной хромосомы. Номер производной хромосомы соответствует номеру интактной хромосомы, имеющей ту же центромеру, что и хромосома-дериват:
46,XY,der(9)del(9)(p12)del(9)(q31)
46,XY,der(9) (:р12->q31:)
Дериватная хромосома 9 является результатом двух терминальных делеций, произошедших в коротком и длинном плечах, с точками разрыва в полосах 9р12 и 9q31 соответственно.
46,XX,der (5)add(5)(p15.1)del(5)(q13)
46,XX,der(5)(?::p15.1-»q13:)
Дериватная хромосома 5 с дополнительным материалом неизвестного происхождения, присоединенным к полосе 5р15.1, и терминальной делецией длинного плеча дистальнее полосы 5q13.
Дицентрические хромосомы. Символ die используют для описания дицентрических хромосом. Дицентрическая хромосома заменяет одну или две нормальные хромосомы. Таким образом, нет необходимости указывать недостающие нормальные хромосомы.
45,XX,dic(13;13)(q14;q32)
45,XX,dic(13;13)(13pter->13ql4::13q32-»13pter)
Разрыв и воссоединение произошли в полосах 13ql4 и 13q32 на двух гомологичных хромосомах 13, в результате чего образовалась дицентрическая хромосома.
Дупликации. Дупликации обозначают символом dup; они могут быть прямыми и инвертированными.
46,XX,dup(1)(q22q25)
46,XX,dup(1)(pter->q25::q22->qter)
Прямая дупликация сегмента между полосами lq22 и lq25.
46,XY,dup(1)(q25q22)
46,XY,dup(1) (pter->q25::q25->q22::q25->qter) или (pter->q22::q25-»q22::q22->qter)
Инвертированная дупликация сегмента между полосами lq22 и lq25. Необходимо отметить, что только детальная система дает возможность описать инвертированную дупликацию.
Инверсии. Символ inv используют для описания пара- и перицентрических инверсий.
46,XX,inv(3)(q21q26.2)
46,XX,inv(3)(pter->q21::q26.2->q21::q26.2->qter)
Парацентрическая инверсия, при которой разрыв и воссоединение произошли в полосах 3q21 и 3q26.2 длинного плеча хромосомы 3.
46,XY,inv(3)(p13q21)
46,XY,inv(3)(pter-»pl3::q21->p13::q21->qter)
Перицентрическая инверсия, при которой разрыв и воссоединение произошли между полосой 3р13 короткого плеча и полосой 3q21 длинного плеча хромосомы 3. Участок между этими полосами, включающий центромеру, перевернут на 180°.
Инсерции. Символ ins используют для обозначения прямой или инвертированной инсерции. Прямой считают такую инсерцию, при которой проксимальный конец района вставки оказывается в проксимальном положении относительно второго ее конца. При инвертированной инсерции проксимальный конец района вставки оказывается в дистальном положении. Тип инсерции (прямая или инвертированная) также может быть отражен символами dir и inv соответственно.
46,XX,ins(2)(pl3q21q31)
46,XX,ins(2)(pter->p13::q31->q21::pl3-»q21::q31-qter)
Прямая инсерция, т. е. dir ins(2) (p13q21q31), произошла между сегментами 2q21 и 2q31 длинного плеча и сегментом 2р13 короткого плеча хромосомы 2. Участок хромосомы длинного плеча между сегментами 2q21 и 2q31 вставлен в короткое плечо в районе сегмента 2р13. В новом положении сегмент 2q21 остается ближе к центромере, чем сегмент 2q31.
46,XY,ins(2) (pl3q31q21)
46,XY,ins(2)(pterH>pl3::q21->q31::pl3->q21::q31-»qter)
В данном случае вставленный участок инвертирован, т. е. inv ins(2)(p13q31q21). Во вставке сегмент 2q21 отстоит от центромеры дальше, чем сегмент 2q31. Таким образом, изменилось расположение сегментов по отношению к центромере.
Изохромосомы. Символ i используют при описании изохромосом, которые представляют собой хромосомы, состоящие из двух идентичных плеч. Точки разрыва в изохромосомах локализованы в центромерных районах р10 и q10.
46,XX,i(17)(q10)
46,XX,i(17)(qter-»q10::q10 ->qter)
Изохромосома по длинному плечу хромосомы 17 и точка разрыва обозначены в районе 17q10. В кариотипе одна нормальная хромосома и одна перестроенная хромосома 17.
46,X,i(X)(q10)
46,X,i(X) (qter-»q10::q10->qter)
Одна нормальная Х-хромосома и Х-изохромосома по длинному плечу.
Ломкие участки (fragile sites, сокращенно fra) могут проявляться как нормальный полиморфизм, а могут быть связаны с наследственными заболеваниями или аномалиями фенотипа.
46,X,fra(X)(q27.3)
Ломкий участок в субполосе Xq27.3 одной из Х-хромосом в женском кариотипе.
46,Y,fra(X)(q27.3)
Ломкий участок в субполосе Xq27.3 Х-хромосомы в мужском кариотипе.
Маркерная хромосома (таг) - это структурно измененная хромосома, ни одна часть которой не может быть идентифицирована. Если какая-нибудь из частей аномальной хромосомы идентифицирована, ее описывают как производную хромосому (der). При описании кариотипа перед символом mar ставят знак (+).
47,XX,+mar
Одна дополнительная маркерная хромосома.
48,X,t(X;18)(p11.2;q11.2)+2mar
Две маркерные хромосомы в дополнение к транслокации t(X;18).
Кольцевые хромосомы обозначают символом г, они могут состоять из одной или нескольких хромосом.
46,XX,r(7)(p22q36)
46,XX,r(7) (::р22->q36::)
Разрыв и воссоединение произошли в сегментах 7р22 и 7q36 с потерей участков хромосомы, расположенных дистальнее этих точек разрыва.
Если центромера кольцевой хромосомы неизвестна, но известны сегменты хромосом, содержащихся в кольце, кольцевые хромосомы определяются как производные (der).
46,XX,der(1)r(1;3)(p36.1q23;q21q27)
46,XX,der(1)(::lp36.1->1q23::3q21->3q27::)
Транслокации. Реципрокные транслокации
Для описания транслокаций (t) используют те же принципы и правила, что и для описания других хромосомных перестроек. Для того чтобы отличить гомологичные хромосомы, один из гомологов может быть подчеркнут одинарным подчеркиванием (_).
46,XY,t(2;5)(q21;q31)
46,XY,t(2;5)(2pter2q21::5q31-> 5qter;5pter 5q31::2q21->2qter)
Разрыв и воссоединение произошли в сегментах 2q21 и 5q31. Хромосомы обме-нялись участками, дистальными по отношению к этим сегментам. Первой указывают хромосому с меньшим порядковым номером.
46,X,t(X;13)(q27;ql2)
46,X,t(X;13)(Xpter->Xq27::13ql2->13qter;13pter->3q 12::Xq27->Xqter)
Разрыв и воссоединение произошли в сегментах Xq27 и 13q12. Сегменты, дистальные по отношению к этим участкам, поменялись местами. Поскольку в транслокации участвует половая хромосома, ее записывают первой. Отметим, что правильная запись следующая - 46,X,t(X;13), а не 46,XX,t(X;13).
46,t(X;Y) (q22;q1, 1.2)
46,t(X;Y)(Xpter->Xq22::Yq11.2->Yqter;Ypter->Yq11.2::Xq22->Xqter)
Реципрокная транслокация между X- и Y-хромосомами с точками разрыва Xq22 и Yq11.2.
Транслокации с вовлечением в них целых хромосомных плеч могут быть записаны с указанием точек разрыва в центромерных районах р10 и q10. При сбалансированных транслокациях точку разрыва в половой хромосоме или в хромосоме с меньшим порядковым номером обозначают р10.
46,XY,t(4;3)(p10;q10)
46,XY,t(1;3)(lpteMlpl0::3ql0->3qter;3pter->3p40::4q40->4qter)
Реципрокная транслокация целых хромосомных плеч, при которой короткие плечи хромосомы 1 присоединились к центромере с длинными плечами хромосомы 3, а длинные плечи хромосомы 1 присоединились к коротким плечам хромосомы 3.
При несбалансированных транслокациях целых хромосомных плеч перестроен-ная хромосома обозначается как производная (der) и замещает две нормальные хромосомы.
45,XX,der(1;3) (p10;q10)
45,XX,der(1;3)(1pter->1p10::3q10->3qter)
Производная хромосома, состоящая из короткого плеча хромосомы 1 и длинного плеча хромосомы 3. Недостающие хромосомы 1 и 3 не обозначены, так как они заменены производной хромосомой. Кариотип, таким образом, содержит одну нормальную хромосому 1, одну нормальную хромосому 3 и производную хромосому der(l;3).
Робертсоновские транслокации
Это особый тип транслокаций, возникающих в результате центрического слияния длинных плеч акроцентрических хромосом 13-15 и 21-22 с одновременной потерей коротких плеч этих хромосом. Принципы описания несбалансированных транслокаций, затрагивающих целые плечи, применимы и для описания робертсоновских транслокаций с использованием символа (der). Символ rob также может быть использован при описании этих транслокаций, но его нельзя применять в описании приобретенных аномалий. Точки разрывов хромосом, участвующих в транслокации, указывают в районах q10.
45,XX,der(13;21) (q10;q10)
45,XX,rob(13;21) (q10;q10)
Разрыв и воссоединение произошли в сегментах 13q10 и 21q10 центромерных районов хромосом 13 и 21. Производная хромосома заменила одну хромосому 13 и одну хромосому 21. Нет необходимости указывать недостающие хромосомы. Кариотип содержит одну нормальную хромосому 13, одну нормальную хромосому 21 и der (13;21). Дисбаланс возникает за счет потери коротких плеч хромосом 13 и 21.
23 марта 2015Крупнейшая генетическая лаборатория США Reprogenetics, в соавторстве с ведущими учеными из Китая, ряда Нью-Йоркских институтов и медицинских центов, специализирующиеся в области ПГД, опубликовали результаты новых исследований, в которых утверждается, что мутации могут быть обнаружены в эмбрионах после экстракорпорального оплодотворения (ЭКО).
Для проведения исследования достаточно небольшой (щадящей) биопсии, всего около 10 клеток эмбрионов, при этом большинство новых (De Novo) мутаций, которые вызывают непропорционально высокий процент генетических заболеваний, могут быть обнаружены с помощью ПГД. Уникальность метода заключается в разработке нового оригинального процесса скрининга расширенного целого генома.
Новые (De Novo) мутации происходят только в половых клетках и у эмбрионов после оплодотворения. Как правило, эти мутации не присутствуют в крови родителей и даже всеобъемлющий скрининг родителей-носителей не сможет их выявить. Стандартный ПГД не может обнаружить эти мутации, потому что тесты недостаточно чувствительны или акцентированы только на очень узких специфических участках генома.
"Эти результаты являются важным шагом в развитии скрининга целого генома, направленного на поиск самых здоровых эмбрионов при проведении ПГД", говорит Сантьяго Мане (Santiago Munné), доктор философии, основатель и директор Reprogenetics и основатель Recombine. "Этот новый подход может обнаружить почти все изменения генома, и тем самым устранить необходимость дальнейшего генетического тестирования во время беременности или после рождения, обеспечивая при этом выбор самого здорового эмбриона для передачи будущей матери."
Также научно подтверждено, что новый метод снижает в 100 раз частоту ошибок (по сравнению с предыдущими методами).
"Замечательно, что новые (De Novo) мутации могут быть обнаружены с такой высокой чувствительностью и исключительно низким уровнем ошибок, используя малое число эмбриональных клеток," говорит Брок Питерс, доктор философии и ведущий ученый в проведенном исследовании. "Разработанный метод эффективен не только с медицинской точки зрения, но и с экономической и мы с нетерпением ждем продолжения наших научно-исследовательских работ в этой области."
Новые мутации могут привести к серьезным врожденным расстройствам мозга, таким как аутизм, эпилептические энцефалопатии, шизофрения и другие. Так как эти мутации являются уникальными для конкретного сперматозоида и яйцеклетки, которые участвуют в создании эмбриона, генетический анализ родителей не в состоянии их обнаружить.
"До пяти процентов новорожденных страдают от заболеваний, вызванных генетическим дефектом," говорит Алан Беркли, доктор медицинских наук, профессор, директор департамента акушерства и гинекологии Университета Нью-Йорка Fertility Center. "Наш подход всеобъемлющий и направлен на выявление абсолютно здоровых эмбрионов. Это может значительно облегчить некоторые факторы эмоционального и физического стресса ЭКО, особенно для пар, подверженных риску передачи генетических нарушений."
Статья переведена специально дял программы Школа ЭКО, по материалам
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава РФ, г. Москва Ключевые слова
: дети, синдром Нунан, диагностика.
Key words
: children, syndrome Noonan, diagnostics.
В статье описан синдром Нунан (синдром Ульриха – Нунан, тернероидный синдром с нормальным кариотипом) – редкая врожденная патология, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадические случаи. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского – Тернера у особей женского и мужского пола с нормальным кариотипом. Представлено клиническое наблюдение. Показаны сложности дифференциальнодиагностического поиска, недостаточная информированность клиницистов о данном синдроме и важность междисциплинарного подхода.
Исторические факты
Впервые о необычном синдроме упомянул О. Kobylinski в 1883 году (фото 1).
Старейший известный клинический случай синдрома Нунан, описан в 1883 году О. Kobylinski
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти пациентах со стенозом клапана легочной артерии, малым ростом, гипертелоризмом, умеренным снижением интеллекта, птозом, крипторхизмом и скелетными нарушениями. Доктор Нунан, практиковавшая как детский кардиолог в университете Айовы, заметила, что у детей с редким типом порока сердца – стенозом клапана легочной артерии – часто наблюдались типичные физические аномалии в виде низкого роста, крыловидной шеи, широко посаженных глаз и низко расположенных ушей. Мальчики и девочки поражались одинаково. Доктор Джон Опиц, бывший студент Нунан, первым ввел в употребление термин «синдромом Нунан» для характеристики состояния детей, у которых отмечались признаки, похожие на описанные Нунан. Позже Нунан написала статью «Гипертелоризм с фенотипом Тернера», и в 1971 году на симпозиуме сердечнососудистых заболеваний название «синдром Нунан» стало официально признанным .
Этиология и патогенез
Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью (рис. 1). Ген синдрома Нунан локализован на длинном плече хромосомы 12 . Не исключена генетическая гетерогенность синдрома. Описаны спорадические и семейные формы синдрома с аутосомно-доминантной формой наследования. В семейных случаях мутантный ген наследуется, как правило, от матери, так как из-за тяжелых пороков развития мочеполовой системы мужчины с этим заболеванием часто бесплодны. Большинство описанных случаев являются спорадическими, вызванными мутациями de novo.
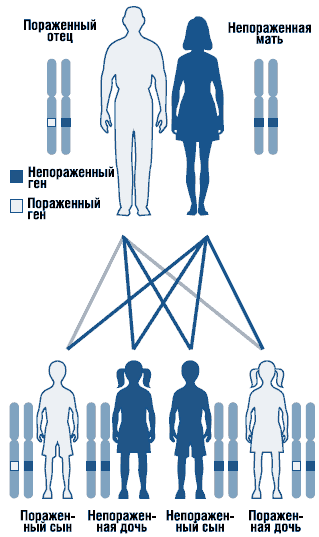
. Аутосомно-доминантный тип наследования
Описанные сочетания синдрома Нунан с нейрофиброматозом I типа в нескольких семьях заставило предположить возможную связь двух независимых локусов 17q11.2 хромосомы 17. У некоторых больных выявляются микроделеции в локусе 22q11 хромосомы 22; в этих случаях клинические проявления синдрома Нунан сочетаются с гипофункцией тимуса и синдромом Ди Джорджи. Ряд авторов обсуждают участие в патогенезе синдрома предполагаемых генов лимфогенеза в связи с наличием сходных с синдромом Тернера лицевых и соматических аномалий и высокой частоты патологии лимфатической системы .
Наиболее частая причина синдрома Нунан – мутация гена PTPN11, которая обнаруживается приблизительно у 50% больных. Белок, кодируемый геном PTPN11, относится к семейству молекул, регулирующих ответ эукариотических клеток на внешние сигналы. Наибольшее число мутаций при синдроме Нунан локализовано в экзонах 3,7 и 13 гена PTPN11, кодирующих домены белка, отвечающие за переход протеина в активное состояние .
Возможные представления о патогенезе представлены следующими механизмами:
RAS-MAPK-путь – очень важный путь сигнальной трансдукции, через который внеклеточные лиганды – определенные факторы роста, цитокины и гормоны – стимулируют клеточную пролиферацию, дифференцирование, выживаемость и метаболизм (рис. 2). После связывания лиганда рецепторы на поверхности клеток фосфорилируются в местах их эндоплазматического региона. Это связывание задействует адаптерные протеины (например, GRB2), которые формируют конститутивный комплекс с факторами обмена гуаниновых нуклеотидов (например, SOS), конвертирующих неактивный ГДФ-связанный RAS в его активную ГТФ-связанную форму. Активированные RAS-протеины затем активируют RAF-MEKERKкаскад через ряд реакций фосфорилирования. В результате активированный ERK проникает в ядро для изменения транскрипции целевых генов и корректирует активность эндоплазматических мишеней для индукции адекватных кратковременных и длительных клеточных ответов на стимул. Все гены, вовлеченные в синдром Нунан, кодируют интегральные для этого пути протеины, и мутации, вызывающие болезнь, обычно усиливают сигнал, проходящий через этот путь.

. RAS-MAPK-сигнальный путь. Ростовые сигналы передаются с активированных фактором роста рецепторов к ядру. Мутации в PTPN11, KRAS, SOS1, NRAS и RAF1 ассоциированы с синдромом Нунан, а мутации в SHOC2 и CBL ассоциированы с подобным синдрому Нунан фенотипом
Клиническая характеристика синдрома Нунан
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с крыловидной складкой или низким ростом волос, низкий рост, гипертелоризм глазных щелей (фото 2). Лицевые микроаномалии включают антимонголоидный разрез глазных щелей, опущенные вниз наружные углы глазных щелей, птоз, эпикантус, низко расположенные ушные раковины, складчатый завиток ушных раковин, аномалии прикуса, расщелину язычка мягкого неба, готическое небо, микрогнатию и микрогению. Грудная клетка щитовидной формы с гипоплазированными широко расставленными сосками, грудина выступает в верхней части и западает в нижней. Около 20% больных имеют умеренно выраженную патологию скелета. Наиболее часто встречаются воронкообразная деформация грудной клетки, кифоз, сколиоз; реже – уменьшение числа шейных позвонков и их сращение, напоминающее аномалии при синдроме Клиппеля – Фейля .

. Фенотипы синдрома Нунан
У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным ростом на темени, часто встречаются пигментные пятна на коже, гипертрихоз, дистрофия ногтевых пластинок, аномалии прорезывания и расположения зубов, склонность к образованию келоидных рубцов, повышенная растяжимость кожи. У трети больных отмечаются периферические лимфатические отеки, чаще лимфедема кистей и стоп проявляется у детей раннего возраста. Нередким признаком является патология зрения (миопия, косоглазие, умеренный экзофтальм и др.). Задержка роста встречается примерно у 75% больных, больше выражена у мальчиков и обычно незначительна. Отставание в росте манифестирует в первые годы жизни, реже отмечается незначительный дефицит роста и массы при рождении. С первых месяцев жизни отмечается снижение аппетита. Костный возраст обычно отстает от паспортного.
Характерным признаком синдрома является одно- или двусторонний крипторхизм, встречающийся у 70–75% больных мужского пола, у взрослых больных отмечается азооспермия, олигоспермия, дегенеративные изменения яичек. Тем не менее пубертат наступает спонтанно, иногда с некоторой задержкой. У девочек часто отмечается задержка становления менструации, иногда – нарушения менструального цикла. Фертильность может быть нормальной у больных обоих полов.
Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто отмечаются особенности поведения, расторможенность, синдром дефицита внимания. Речь обычно развита лучше, чем другие интеллектуальные сферы. Степень снижения интеллекта не коррелирует с тяжестью соматических нарушений [Маринчева Г.С., 1988]. В единичных случаях описываются пороки развития центральной нервной системы (гидроцефалия, спинномозговые грыжи), тромбоэмболические инфаркты мозга, возможно, связанные с гипоплазией сосудов .
Пороки внутренних органов при синдроме Нунан достаточно характерны. Наиболее типичными являются сердечно-сосудистые аномалии: клапанный стеноз легочной артерии (около 60% больных), гипертрофическая кардиомиопатия (20–30%), структурные аномалии митрального клапана, дефекты предсердной перегородки, тетрада Фалло; коарктация аорты описана только у больных мужского пола.
У трети больных регистрируются пороки мочевыделительной системы (гипоплазия почек, удвоение лоханок, гидронефроз, мегауретер и др.).
Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах в ротовой полости и носоглотке. Обнаруживаются различные дефекты коагуляции: недостаточность тромбоцитарной системы, снижение уровня факторов свертывания, особенно XI и XII, увеличение тромбопластинового времени . Имеются сообщения о сочетании синдрома Нунан с лейкемией и рабдомиосаркомой, что может свидетельствовать о некотором повышении риска малигнизации у этих больных .
В таблице 1 представлены особенности фенотипа при синдроме Нунан, меняющиеся с возрастом пациента. В таблице 2 – корреляция между фенотипом и генотипом при синдроме Нунан.
Таблица 1 . Типичные черты лица у больных синдромом Нунан по возрастам
| Лоб, лицо, волосы | Глаза | Уши | Нос | Рот | Шея | |
| Новорожденный* | Высокий лоб, низкая линия роста волос в затылочной области | Гипертелоризм, наклонные книзу глазные щели, складка эпикантуса | – | Короткий и широкий утопленный корень, вздернутый кончик | Глубоко утопленный губной желобок, высокие широкие пики красной каймы губ, микрогнатия | Избыточная кожа на затылке |
| Грудной (2–12 мес.) | Большая голова, высокий и выпирающий лоб | Гипертелоризм, птоз или толстые нависающие веки | – | Короткий и широкий утопленный корень | – | – |
| Ребенок (1–12 лет) | Грубые черты, вытянутое лицо | – | – | – | – | – |
| Подросток (12–18 лет) | Миопатичес-кое лицо | – | – | Мостик высокий и тонкий | – | Очевидное формирование шейных складок |
| Взрослый (>18 лет) | Отличительные черты лица утонченные, кожа кажется тонкой и прозрачной | – | – | Выпирающая носогубная складка | – | – |
| Все возрасты | – | Голубые и зеленые радужные оболочки, ромбовидные брови | Низкие, ротированные назад уши с толстыми складками | – | – | – |
Таблица 2 . Корреляции между генотипом и фенотипом при синдроме Нунан*
| Сердечнососудистая система | Рост | Развитие | Кожа и волосы | Другое | |
| PTPN11 (примерно 50%) | Более выражен стеноз легочного ствола; меньше – гипертрофическая кардиомиопатия и дефект межпредсердной перегородки | Более низкий рост; ниже концентрация IGF1 | Пациенты с N308D и N308S имеют слабое снижение или нормальный интеллект | – | Больше выражен геморрагический диатез и ювенильная миеломоноцитарная лейкемия |
| SOS1 (примерно 10%) | Меньше дефект межпредсердной перегородки | Более высокий рост | Меньше снижение интеллекта, задержка развития речи | Подобны сердечно-кожно-лицевому синдрому | – |
| RAF1 (примерно 10%) | Больше тяжелая гипертрофическая кардио-миопатия | – | – | Больше родимых пятен, лентиго, пятен кофе с молоком | – |
| KRAS (<2%) | – | – | Более тяжелая задержка когнитивного развития | Подобны сер-дечно-кожно-лицевому синдрому | – |
| NRAS (<1%) | – | – | – | – | – |
Данные лабораторных и функциональных исследований
Специфических биохимических маркеров для диагностики синдрома Нунан не существует. У некоторых больных выявляется снижение спонтанной ночной секреции гормона роста при нормальном ответе на фармакологические стимулирующие тесты (клофелином и аргинином), снижение уровня соматомедина-С и снижение реакции соматомединов на введение гормона роста.
Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях диагноз подтверждается результатами молекулярно-генетического исследования. Критерии диагностики синдрома включают наличие характерного лица (при нормальном кариотипе) в сочетании с одним из следующих признаков: патологии сердца, низкий рост или крипторхизм (у мальчиков), задержка полового созревания (у девочек). Для выявления сердечно-сосудистой патологии необходимо проведение ультразвукового исследования сердца с динамическим определением размеров полостей и стенки желудочков. Возможна пренатальная диагностика заболевания при помощи ультразвукового мониторинга, позволяющего выявить пороки сердца и аномалии строения шеи .
Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера; уточнить диагноз позволяет цитогенетическое исследование. Фенотипические признаки синдрома Нунан встречаются при ряде других заболеваний: синдроме Вильямса, синдроме LEOPARD, Дубовица, кардиофацио-кожном синдроме, Корнелии де Ланге, Коэна, Рубинштейна – Тейби и др. Точная идентификация этих заболеваний будет возможна только при проведении молекулярногенетических исследований каждого синдрома при значительном клиническом материале, что в настоящее время активно развивается.
Лечение
Лечение больных с синдромом Нунан направлено на устранение пороков сердечно-сосудистой системы, нормализацию психических функций, стимуляцию роста и полового развития. Для лечения больных с дисплазией клапанов легочной артерии, помимо прочих методов, с успехом применяется баллонная вальвулопластика. С целью стимуляции психического развития применяются ноотропные и сосудистые средства. Препараты, направленные на стимуляцию полового развития, показаны в основном больным с крипторхизмом. Применяются препараты хорионического гонадотропина в возрастных дозировках. В старшем возрасте – при наличии гипогонадизма – препараты тестостерона. В последние годы применяются рекомбинантные формы гормона роста человека в лечении больных с синдромом Нунан . Клинические данные подтверждаются увеличением на фоне терапии уровня соматомедина-С и специфического связывающего белка. Конечный рост больных, длительное время получающих терапию гормоном роста, в некоторых случаях превышает средний рост членов семьи.
Прогноз для жизни определяется тяжестью сердечно-сосудистой патологии.
Профилактика болезни основывается на данных медико-генетического консультирования.
Медико-генетическое консультирование
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в генах: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разрабатываются способы пренатальной диагностики заболевания.
Клиническое наблюдение
Мальчик Г., 9 лет (фото 3), наблюдался по месту жительства врачом-генетиком с диагнозом «хромосомная патология?, синдром Вильямса (своеобразный фенотип, уплотнение створок митрального клапана, гиперкальциемия однократно в 3 года)?.

. Особенности фенотипа ребенка с синдромом Нунан (удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, нос укорочен с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия)
Жалобы на сниженную память, утомляемость, сниженные темпы роста.
Анамнез семейный : родители русские по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей, здоровые. Рост отца – 192 см, рост матери – 172 см. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии не отмечалось.
Анамнез жизни и заболевания : мальчик от 2-й беременности (1-я беременность – м/а), протекавшей с угрозой прерывания на всем протяжении, сопровождающейся многоводием. Роды первые, в срок, стремительные, масса при рождении – 3400 г, длина – 50 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. При рождении неонатологом обращено внимание на необычный фенотип ребенка, рекомендовано исследование кариотипа, результат – 46, XY (нормальный мужской кариотип). Был заподозрен врожденный гипотиреоз, проведено исследование тиреоидного профиля, результат – нормальный тиреоидный статус. Далее ребенок наблюдался генетиком с предполагаемым диагнозом «синдром Вильямса». Ранний постнатальный период – без особенностей. Моторное развитие по возрасту, первые слова – к году, фразовая речь – в 2 года 3 мес.
В возрасте 8 лет консультирован эндокринологом по поводу сниженных темпов роста, утомляемости, сниженной памяти. При рентгенологическом исследовании кистей рук выявлено умеренное отставание костного возраста (КВ) от паспортного (КВ соответствовал 6 годам). При исследовании тиреоидного профиля выявлено умеренное повышение тиреотропного гормона при нормальном уровне свободного Т4 и остальных показателей; УЗИ щитовидной железы – без патологии. Назначена гормональная терапия с последующим динамическим наблюдением.
Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Московский областной консультативно-диагностический центр для детей с целью уточнения диагноза.
Данные объективного исследования:
Рост – 126 см, вес – 21 кг.
Физическое развитие ниже среднего, гармоничное. Sds роста соответствует –1 (норма – –2+2). Особенности фенотипа (фото 3): удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, низкий рост волос на шее, нос укороченный с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия, гипертелоризм сосков, асимметрия грудной клетки, на стопах неполная кожная синдактилия 2–3-го пальцев, выраженная гипермобильность межфаланговых суставов, ломкие, сухие ногти. По внутренним органам – без особенностей. Половое развитие – Tanner I (что соответствует допубертатному периоду).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови – показатели в пределах нормы.
Тиреоидный профиль (ТТГ) – 7,5 мкМЕ/ мл (норма – 0,4–4,0), остальные показатели в норме.
Соматотропный гормон (СТГ) – 7 нг/мл (норма – 7–10), соматомедин-С – 250 нг/мл (норма – 88–360).
УЗИ щитовидной железы – без патологии.
УЗИ внутренних органов – без особенностей.
ЭКГ – синусовая тахикардия, нормальное положение электрической оси сердца.
ЭхоКГ – ПМК I степени с минимальной регургитацией, миксоматозное утолщение створок митрального клапана, дополнительная хорда в полости левого желудочка.
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника I степени.
R-графия кистей рук с захватом предплечий – костный возраст 7–8 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – без патологических изменений.
Аудиограмма – без патологии.
ДНК-диагностика: молекулярно-генетическое исследование – делеций исследуемых локусов критического района хромосомы 7 не выявлено; обнаружена мутация Gly434Ary (1230G>A) в 11-м экзоне гена SOS1 (анализ гена PTPN11 – мутаций не обнаружено), что характерно для синдрома Нунан.
Консультации специалистов:
Эндокринолог – субклинический гипотиреоз, неполная медикаментозная компенсация.
Окулист – астигматизм.
Невролог – вегетососудистая дистония. Невротические реакции.
Кардиолог – функциональная кардиопатия.
Хирург-ортопед – нарушение осанки. Деформация грудной клетки.
Генетик – синдром Нунан.
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований, поставлен диагноз «синдром Нунан», что подтверждено результатом молекулярно-генетического исследования.
Таким образом, представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний, важность молекулярно-генетических методов для уточнения диагноза. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждению повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии.
Список литературы:
- Baird P., De Jong B. Noonan’s syndrome (XX and XY Turner phenotype) in three generations of a family // J. Pediatr., 1972, vol. 80, p. 110–114.
- Hasegawa T., Ogata T. et al. Coarctation of the aorta and renal hupoplasia in a boy with Turner/Noonan surface anomalies and a 46, XY karyotype: a clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata // Hum. Genet., 1996, vol. 97, р. 564–567.
- Федотова Т.В., Кадникова В.А. и соавт. Клинико-молекулярно-генетический анализ синдрома Нунан. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика, приложение к № 5, 2010, с.184.
- Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? // Br. J. Dermatol., 1994, vol. 131, р. 270–274.
- Municchi G., Pasquino A.M. et al. Growth hormone treatment in Noonan syndrome: report of four cases who reached fi nal height // Horm. Res., 1995, vol. 44, р. 164–167.
Материал из BrainstormWiki
Что такое мутация?
Начнем с того, что “правильного” генокода не существует. Поэтому если у меня ген один, а у вас другой, то это не означает еще, что один из нас мутант.
Тем не менее, существуют такие изменения в генокоде, которые приводят к явным проблемам, и обычно они редко встречаются; их и называют мутации. Изменения, которые встречаются у более чем 1% населения, правильнее называть не мутации, а полиморфизмы .
Мутации можно унаследовать у родителей, или же они могут возникнуть только у ребенка - в этом случае их называют de novo
Мутации могут возникнуть уже при развитии организма, при делении и дифференциации клеток - такие называют соматическими мутациями ; их нельзя унаследовать, потому что они “живут” в клетках тела (сома), но не в половых клетках..
Наоборот, мутации, которые затрагивают половые клетки, наследуются, и их называют мутациями в зародышевой линии (germline mutations)
Мутации могут быть молчаливыми - они есть, а эффекта нет. Таких подавляющее большинство - если помните, гены занимают лишь менее 2% всей ДНК. Каждый из нас является носителем как минимум десятков таких мутаций. Если они приходятся на малозначимые участки ДНК, никто ничего не заметит.
Мутации могут затрагивать одну “букву” генокода - а могут и целый огромный фрагмент ДНК. Этот фрагмент чаще всего может быть потерян (делеция) или задвоен (дупликация) - в этом случае клетки изменят количество синтезируемых белков по затронутым генам - изменится их доза
Для разных видов мутаций существуют разные виды анализов и тестов, и мы по ним сейчас попробуем пройтись.
От больших - к маленьким
Анеуплодия - аномалия числа хромосом
Самая большая по размеру генетическая аномалия - изменение числа хромосом. Хотя это и не аутизм, но начать стоит с него 50 лет назад было открыто, что синдром Дауна вызывается тремя 21 хромосомами, вместо двух. Это называется трисомией; а изменение вообще числа хромосом - анеуплодией. Помимо этого встречается трисомия 13 хромосомы (синдром Патау) и 18 (синдром Эдвардса), а также ряд анеуплодий половых хромосом (X и Y)
Все это можно разглядеть в обычный оптический микроскоп - анализ называется “кариотипирование”. Хромосомы делящихся клеток фотографируют, сортируют и описывают. На картинке виден кариотип женщины с тремя 21 хромосомами.
Эффект от трисомий огромен и заметен в очень многих процессах: и в строении тела, и в физиологических процессах, и даже по характерным белкам в крови носящей матери (именно так работают сейчас скрининговые тесты на с. Дауна)
Почему встречаются только трисомии 21, 13 и 18 и половых хромосом? Случиться это может с любой хромосомой, но остальные не выживают даже до стадии заметной беременности. Возможно причина в том, что 21, 13 и 18 хромосомы - одни из самых “бедных” белковыми генами (помните, нумерация там по размеру, они ведь еще и маленькие) и увеличение их дозы относительно переносимо. Это подтверждается таблицей ниже: на ранних фазах развития возможны любые анеуплодии, а по мере приближения к успешным родам - только вот эти 3.

Copy Number Variation

CNV - это тоже либо задвоение, либо отсутствие определенной части ДНК, но уже не на уровне целой хромосомы, а несколько миллионов нуклеотидов.
Для их обнаружения применяют тестирование, называемое CGH array: comparative genomic hybridization, также FISH и другие.В нашей практике часто называют также “молекулярное кариотипирование”, микроматричный хромосомный анализ и т.п.
Это все равно огромные куски ДНК, которые могут включать в себя десятки генов и управляющих структур. Эффект от них очень заметен, почти всегда включает в себя дисморфизмы и измененения когнитивных способностей. Обозначают их по затронутому участку генокода, например 2q32 deletion - потеря участка в полоске 32 на длинной руке 2 хромосомы (вспоминаем часть 1 и “адреса” участков хромосом) Туда же относятся и многие известные “именные” синдромы, например синдром Вильямса - 7q11.23 deletion
Такие CNV мутации, как считается объясняют от 3% до 10% случаев аутизма. Это практически единственный вид генетической аномалии, который уверенно связан с некоторыми - синдромными - формами аутизма.
Еще раз - основное, что нужно знать про CNV в аутизме: они работают в небольшой доле случаев,но их эффект заметен в очень многих аспектах, от дисморфизмов до интеллекта. Т.е. наличие дефектов в CNV почти всегда можно увидеть либо прямо на лице, либо в виде довольно очевидных нарушений типа системной гипотонии и атаксии...
Некоторые известные и документированные CNV

При этом наличие данных CNV не гарантирует аутизма как диагноза. Пенетрантность не 100% никогда. Недавнее эстонское исследование группы людей без каких-либо диагнозов показало, что носители CNV 16p11.2 демонстрируют диcморфизмы головы, ожирение, нарушение когнитивных способностей - но не имеют диагноза аутизма, вопреки данным в таблице выше;
Существует онлайн-база данных CNV, вызывающих аутизм и сходные синдромы http://projects.tcag.ca/autism/
Синдромный аутизм
Синдромным аутизм называют тогда, когда считается, что аутистические симптомы вызваны понятным генетическим расстройством - как правило CNV, но не только (см. Fragile X например). Многие ученые говорят, что термин этот некорректный. Правильнее наверное говорить "аутизм понятной генетической этиологии" или что-нибудь в этом роде.
Очень хороший обзор самых известных форм аутизма с "понятной генетической этиологией" - т.е. синдромных форм аутизма можно найти в блоге Эмили Казанова
- Часть 1: синдромы Тимоти, Smith-Lemli-Opitz, CHARGE, Cornelia de Lange, Lujan-Fryns
Еще раз про пенетрантность
Пенетрантность - penetrance - это термин, означающий процент носителей мутации, у которой проявляется (в данном случае) аутизм. В обзоре в предыдущей главе видно, что пенетрантность для синдромных форм аутизма от 60 до 90 процентов
Пенетрантность в 92% для idic(15) называют "ошеломительной". Да, это много. Это более чем достаточно, чтобы считать данный генетический вариант причиной аутизма - если он найден у ребенка и симптомы сходятся. Это же дает основания полагать, что если эту мутацию исключить при PGD - у следующего ребенка тех же родителей аутизма не будет.
SNP

Теперь давайте доберемся до самых маленьких генетических вариаций, заключенных всего в одном нуклеотиде, одной “букве” В человеческом геноме есть несколько миллионов точек, где распространены вариации в одной “букве” генокода; вокруг все стабильно, а вот в этих конкретных местах - у разных людей по разному. Причем такие вариации почему-то почти всегда биаллельны, то есть там только два варианта “букв” из четырех, и присутствуют они у очень большой доли населения - часто один вариант у 60%, а другой - у 40%. Это не мутация, а полиморфизм, если вспомнить часть 7. (для того, чтобы SNP официально признали таковым, его распространенность должна быть не менее 1%)
То есть это не “мутации”, не ошибки ДНК . Это норма и ее варианты.
Такие вариации называют SNP (читается “снип”), обозначают номерами типа rs2320030 и их найдено и описано порядка 10 миллионов.
Genome-wide association studies
Анализ, показывающий какие варианты (аллели) SNP присутствуют у конкретного человека проводят на SNP чипе. Он представляет из себя пластинку, на которой “напечатаны” образцы генома человека вокруг популярных SNP, ДНК испытуемого (часто образец слюны) наносится на эту пластину и далее смотрят, куда она “прилипла”, а куда нет. Популярный анализ 23andme именно такой, и дает около 1 миллиона таких SNP. Это называется “генотипирование” и оно довольно дешево, а поэтому его применяют сейчас часто (и часто не по делу). Дешево потому, что SNP хорошо известны, и этот анализ удобно делать на потоке, массово.
Эта доступность породила особый вид исследований, Genome-Wide Association Studies (GWAS), при которых берется группа носителей наследуемого признака, и контрольная группа, всех генотипируют, а затем смотрят, нет ли какого-нибудь SNP, который бы у первых был в одном варианте, а у вторых - в другом.
Проблема здесь в статистике: так как SNPов миллионы, велика вероятность того, что мы увидим связь там, где её нет: просто например, на группе в 30 аутистов найдется такой SNP который 30 раз выпадет “правильно” у них. Если монетку бросить миллион раз, она и на ребро встанет:) С этой проблемой можно бороться статистическими же методами, и бороться успешно. Однако в целом GWAS результаты для аутизма отвратительно реплицируются.
Что означает наличие SNP

Эффект от одного или другого варианта SNP как правило мизерный. По другому и быть не может, так как они все по определению очень распространены в популяции. Однако есть заболевания (например муковисцидоз) для которых определяющими являются именно они (но как правило не в одном SNP: обычно наличие “плохого” варианта статистически повышает риск расстройства, скажем, на 2%)
Ни одного SNP, связанного хоть сколь-нибудь значимо с аутизмом, не найдено. Кстати, те, что связаны хоть сколько-нибудь - как правило, сидят не в генах, а в управляющих элементах ДНК - т.е. не в экзоме.
Поэтому все тесты, которые используют SNP генотипирование и рассчитывают значимый “риск аутизма” - шарлатанство скорее всего. Есть не мало сайтов, предлагающих загрузить результаты 23andme и получить “персональные” рецепты лечения аутизма. Самый знаменитый - это “доктор” Yasko, продвигающий рецепты от MTHFR “мутаций”. Шарлатанство. Еще раз, это не мутации. MTHFR - важный ген в фолатном обмене, и он вероятно связан с аутизмом, но не на SNP уровне это искать.
Справедливости ради надо сказать, что комбинация SNP (сотен разных снипов, каждый из которых дает мизерный вклад, но в сумме набирается что-то существенное) кажется способна объяснить шизофрению (в отличие от аутизма) - что пытаются выявить почти те же люди, что делали Human Genome Project, и почти столь же титаническими усилиями. Но там речь идет именно что о сотне снипов.
De novo мутации

“Настоящие” мутации, не живущие постоянно в популяции, мутации, которые появляются у конкретных людей и передаются их детям (или возникают у детей), называются de novo мутациями. Они могут быть как большими участками (CNV), как маленькими вставками и удалениями (indels) так и отдельными “буквами”. Этот пост про последние два вида.
Для их обнаружения тесты на дешевых чипах непригодны, геном надо читать подряд, или, как это называется, секвенировать (sequencing). Можно читать как весь геном вообще - whole genome sequencing (дорого), так и только экзом, только часть, кодирующую белки - exome sequencing, (относительно недорого, $1000 за человека). Соответственно, в клинической практике оно не применяется почти совсем.
Таких de novo мутаций в принципе довольно много - каждый из родителей передает ребенку не менее 100 таких, но только малая часть выпадает на значимые места ДНК. Свежие уважаемые публикации (Iossifov и коллеги 2012) оценивают ключевой вклад de novo мутаций в приблизительно 10% случаев аутизма. Если вы были на конференции в мае, там был доклад Nagwa Meguid о “Новом гене и уникальной форме аутизма” - ровно такой случай, de novo мутации (различные) в гене BCKDK.
Однако и в этой простой истории: вот мутация, вот она затронула ген, важный для работы мозга, вот мы продемонстрировали, что этот ген влияет на аутизм - есть большая загадка. Если, в отличие от Ивана Иосифова исследовать семьи не с одним, а с двумя аутистами, как это сделал Yuen и коллеги в этом году, то оказывается, что они ухитряются брать разные мутации от одних и тех же родителей.
Обобщаем и классифицируем виды “мутаций”
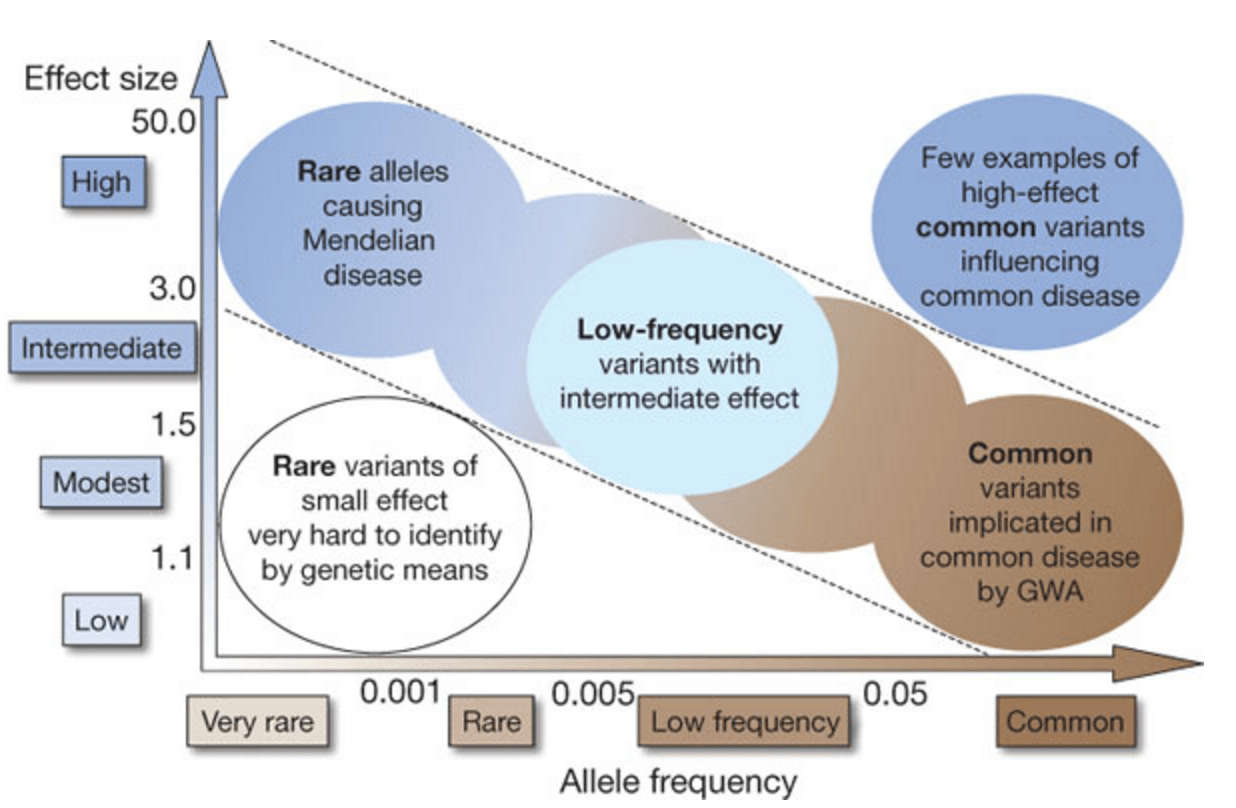
В предыдущих частях мы кратко прошлись по различным видам генетических вариантов в ДНК. От больших по размеру, как правило имеющих заметный эффект, до совсем маленьких.
И правило здесь такое: большие по размеру аномалии как правило редки, но имеют заметный эффект на многие аспекты фенотипа маленькие варианты часто распространены в популяции, но каждая из них имеет мизерный эффект
Это можно суммировать на диаграме типа вот такой, где по горизонтали - распространенность аллели (генетического варианта), от редких к частым, а по вертикали - проявленный эффект у носителей.
Надо отметить, что для аутизма и шизофрении даже самые “сильные” CNV не имеют менеделевского эффекта, обычно повышая риск развития клинически определяемого заболевания с 1% до 3%-10%. Это называется incomplete penetrance . Можно быть носителем известного “шизофренического CNV”, можно иметь дизморфизмы и отклонения в эндокринной системе, характерные для него, но не иметь шизофрении, хотя и обнаруживать некоторые субклинические отклонения.
Соответственно, индивидуальный эффект от полиморфизмов типа SNP вообще мизерный и почти не имеет практического значения для диагностики аутизма.
| Ликбез по генетике аутизма |
|---|
| I. Ликбез по генетике аутизма II. Генетика и близнецы III. Виды мутаций IV. |
Трем группам американских ученых, независимо друг от друга, удалось впервые установить связь между мутациями в определенных генах и вероятностью развития у ребенка расстройств аутистического спектра, сообщает The New York Times. Кроме того, исследователи нашли научное подтверждение ранее выявленной прямой зависимости между возрастом родителей, в особенности отцов, и риском развития аутизма у потомства.
Все три группы сосредоточились на редко встречающейся группе генетических мутаций, названной "de novo". Эти мутации не наследуются, а возникают в процессе зачатия. В качестве генетического материала были взяты образцы крови членов семей, в которых родители не являлись аутистами, а у детей развились различные расстройства аутистического спектра.
Первая группа ученых под руководством Мэтью Стэйта (Matthew W. State), профессора генетики и детской психиатрии из Йэльского университета, чья работа опубликована 4 апреля в журнале Nature, проанализировала наличие мутаций "de novo" у 200 человек с диагнозом "аутизм", чьи родители, братья и сестры не являлись аутистами. В результате были обнаружены два ребенка с одинаковой мутацией в одном и том же гене, при этом их не связывало больше ничего, кроме диагноза.
"Это как при игре дартс дважды попасть дротиком в одну и ту же точку на мишени. Вероятность того, что обнаруженная мутация связана с аутизмом - 99,9999 процентов", - цитирует издание профессора Стэйта.
Команда под руководством Ивэна Эйхлера (Evan E. Eichler), профессора генетики из университета штата Вашингтон, исследуя образцы крови 209 семей, в которых есть дети-аутисты, обнаружила такую же мутацию в том же самом гене у одного ребенка. Кроме того, были выявлены двое детей-аутистов из разных семей, у которых оказались идентичные между собой мутации "de novo", но в других генах. Таких совпадений у испытуемых - не аутистов замечено не было.
Третья группа исследователей, которую возглавлял профессор Марк Дэли (Mark J. Daly) из Гарвардского университета, обнаружила у детей-аутистов несколько случаев мутаций типа "de novo" в тех же трех генах. Хотя бы одна мутация такого типа присутствует в генотипе любого человека, но, полагает Дэли, у аутистов, в среднем, их значительно больше.
Все три группы исследователей также подтвердили и ранее замеченную связь между возрастом родителей и аутизмом у ребенка. Чем старше родители, в первую очередь отец, тем выше риск появления мутаций "de novo". Проанализировав 51 мутацию, команда под руководством профессора Эйхлера выяснила, что в мужской ДНК такого рода повреждения встречаются в четыре раза чаще, чем в женской. И тем чаще, если возраст мужчины превышает 35 лет. Таким образом, предполагают ученые, именно поврежденный отцовский генетический материал, получаемый потомством при зачатии, является источником тех мутаций, которые влекут за собой развитие аутистических расстройств.
Ученые согласны в том, что поиск путей предотвращения такого развития событий будет долгим, исследование генетической природы аутизма только начинается. В частности, команды Эйхлера и Дэли нашли свидетельства того, что гены, в которых обнаружены мутации "de novo", задействованы в одних и тех же биологических процессах. "Но это лишь верхушка верхушки айсберга, - считает профессор Эйхлер. - Главное, что все мы сошлись на том, с чего надо начать".
Похожие записи:
 Анализ стихотворения «Поет зима – аукает С есенин идет зима аукает жанр
Анализ стихотворения «Поет зима – аукает С есенин идет зима аукает жанр
 "Спасибо товарищу Сталину за наше счастливое детство" Спасибо товарищу сталину за наше счастливое детство
"Спасибо товарищу Сталину за наше счастливое детство" Спасибо товарищу сталину за наше счастливое детство
 Жириновский жмет, коммунисты отстают
Жириновский жмет, коммунисты отстают
 История рюриковичей и их генеалогическое дерево
История рюриковичей и их генеалогическое дерево
 Давальческое сырье: бухгалтерский учет Давальческая схема 1с
Давальческое сырье: бухгалтерский учет Давальческая схема 1с





